Gorelov Vitaly
CNRS researcher at École Polytechnique

Research Activity
My general interests lay in understanding and predicting the optical and electronic properties of solids and using this knowledge in the field of photovoltaics. In order to achieve this goal I develop theoretical and numerical approaches based on many-body perturbation theory (MBPT) and quantum Monte Carlo theory (QMC) and use these methods to obtain accurate predictions for excitations in complex materials and to develop theoretical models for large-scale predictions in collaboration with experimentalists.
Publications and Talks
Recent Articles
-
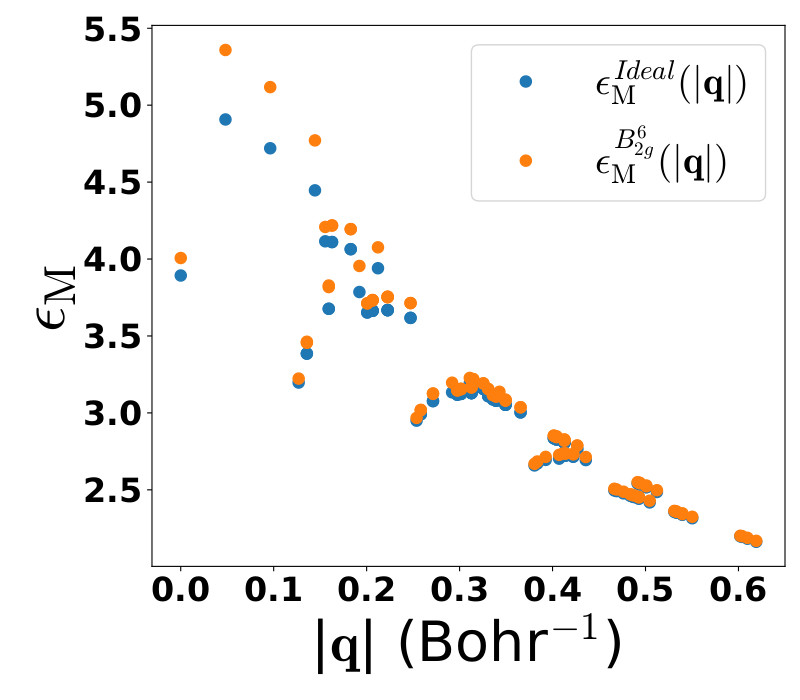
Robustness of electronic screening effects in electron spectroscopies: Example of V2O5
In bulk and low-dimensional extended systems, the screening of excitations by the electron cloud is a key feature governing spectroscopic properties. Widely used computational approaches, such as the GW approximation and the resulting approximate Bethe-Salpeter equation, are explicitly formulated in terms of the Coulomb interaction screned by the macroscopic dielectric function (see Figure). In this work we explore the effect of screening in absorption and electron energy loss spectroscopy, concentrating on the effect of local distortions (along the phonon modes) on the screening and elucidating the resulting changes in the various spectra. Using the layered bulk oxide V2O5 as prototype material, we show in which way local distortions affect the screening, and in which way changes in the screening impact electron energy loss and absorption spectra including excitons. This yields insight concerning the structure-properties relations that are crucial for the use of V2O5 as energy storage material, and more generally, that may be used to optimize the analysis and the calculation of electronic spectra in complex materials.
-
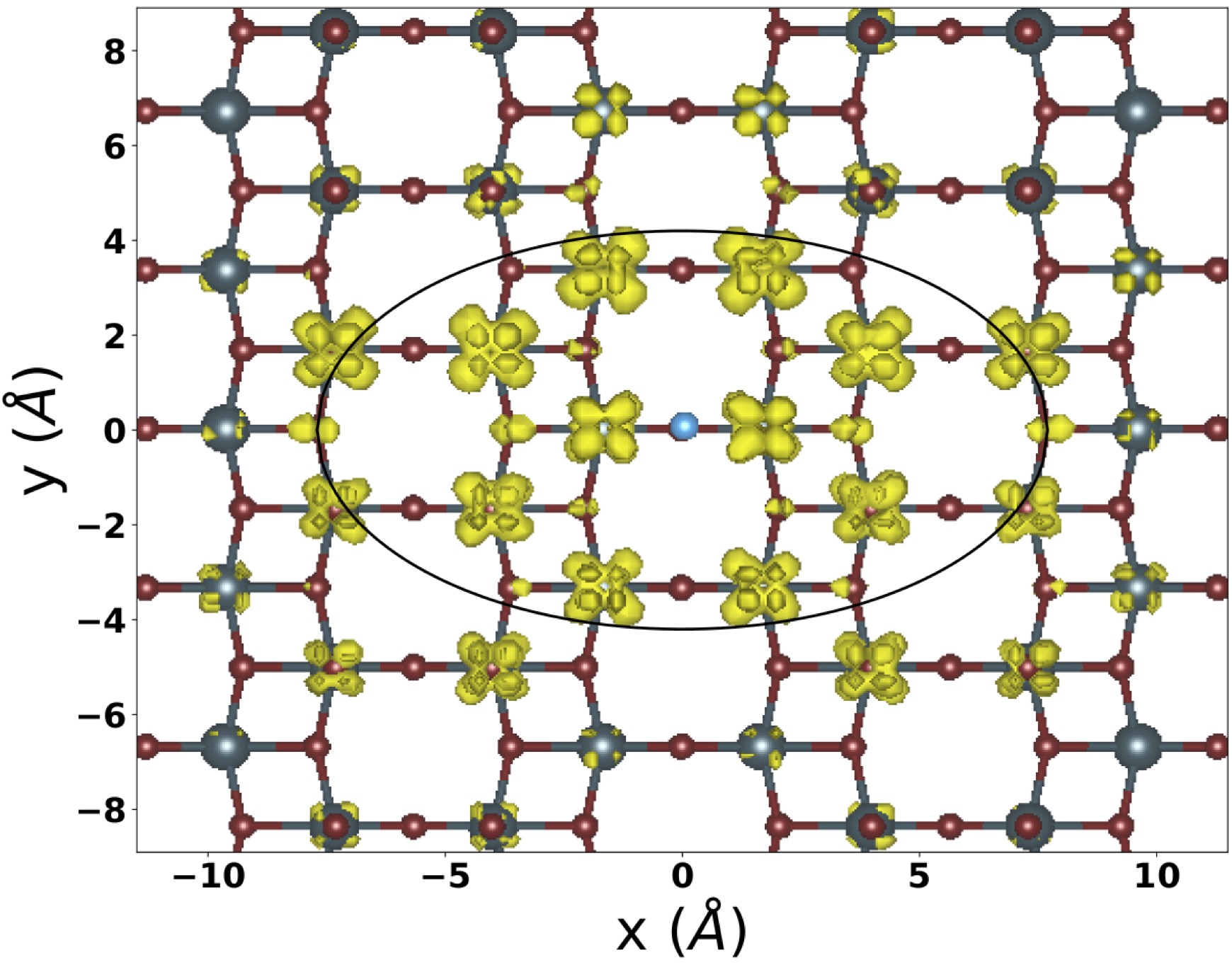
Delocalization of dark and bright excitons in flat-band materials and the optical properties of V2O5
The simplest picture of excitons in materials with atomic-like localization of electrons is that of Frenkel excitons, where electrons and holes stay close together, which is associated with a large binding energy. Here, using the example of the layered oxide V2O5, we show how localized charge-transfer excitations combine to form excitons that also have a huge binding energy but, at the same time, a large electron-hole distance, and we explain this seemingly contradictory finding. The anisotropy of the exciton delocalization is determined by the local anisotropy of the structure, whereas the exciton extends orthogonally to the chains formed by the crystal structure. Moreover, we show that the bright exciton goes together with a dark exciton of even larger binding energy and more pronounced anisotropy. These findings are obtained by combining first principles many-body perturbation theory calculations, ellipsometry experiments, and tight binding modelling, leading to very good agreement and a consistent picture. Our explanation is general and can be extended to other materials.
LANGUAGE SKILLS
-
French Fluent
-
English Fluent
-
Russian Native
Conferences Organized
- The 18th ETSF Young Researchers' Meeting, held in September 2022, Marseille (France). Link here
Annual workshop gathering young researchers in electronic sturcuture and theoretical spectroscopy
Affiliations and details
Laboratoire des Solides Irradies
Theoretical Spectroscopy Group
91128 Palaiseau cedex, France
- vitaly.gorelov@polytechnique.edu
- +33 (0)1 6933 44 91
- gorelov93
- orcID 0000-0003-4324-506X
- Scholar: Vitaly Gorelov
Positions
Studies
Tutoring and Supervision
Awards and Fellowship

Teaching
- Chargé de trava dirigés(TD) et pratiques (TP) (Bachelor level)
Programming and Algorithms in C (TD L1)202330 hElectronics (TD&TP L1)202215 hElectromagnetism (TP L2)20229 hGeometrical Optic (TP L2)20216 hWaves and Vibrations (TD L2)202112 h