
Time-dependent density functionnal theory is an ab initio framework extremely powerfull tool to simulate electronic excitations of materials. Even if it is a longitudinal formalism, it allows the simulation to the absorption spectrum, which is the response to a transverse field. The theory is well established for infinite crystals (3D), where the periodic boundary conditions suggest naturally to work in the reciprocal space, and relies on two results: i) the longitudinal-longitudinal (LL) and transverse-transverse (TT) contraction of the dielectric tensor are equal in the optical limit and ii) the LL contraction is equal to the inverse of the macroscopic average of the inverse dielectric matrix, which is the quantity calculated within TDDFT.
Beyond the bulk materials, some other objects, like surfaces or nano-objects, present interesting electronic properties. These systems, for which the periodicity is partially or totally removed, have been widely studied within the reciprocal space scheme. For this one builds a supercell, which contains a certain amount of matter, and vacuum to isolate the object from the artificial replicas arising from the periodically repeated supercell.
Beyond the bulk materials, surfaces have been widely studied within the reciprocal space scheme. More recently, the promise of new electronic properties expected from nanomaterials has lead to apply this formalism to isolated nano-object. For these systems, for which the periodicity is partially or totally removed, one builds a supercell, which contains a certain amount of matter, and vacuum to isolate the object from the artificial replicas arising from the periodically repeated supercell.
Selected-G formalism
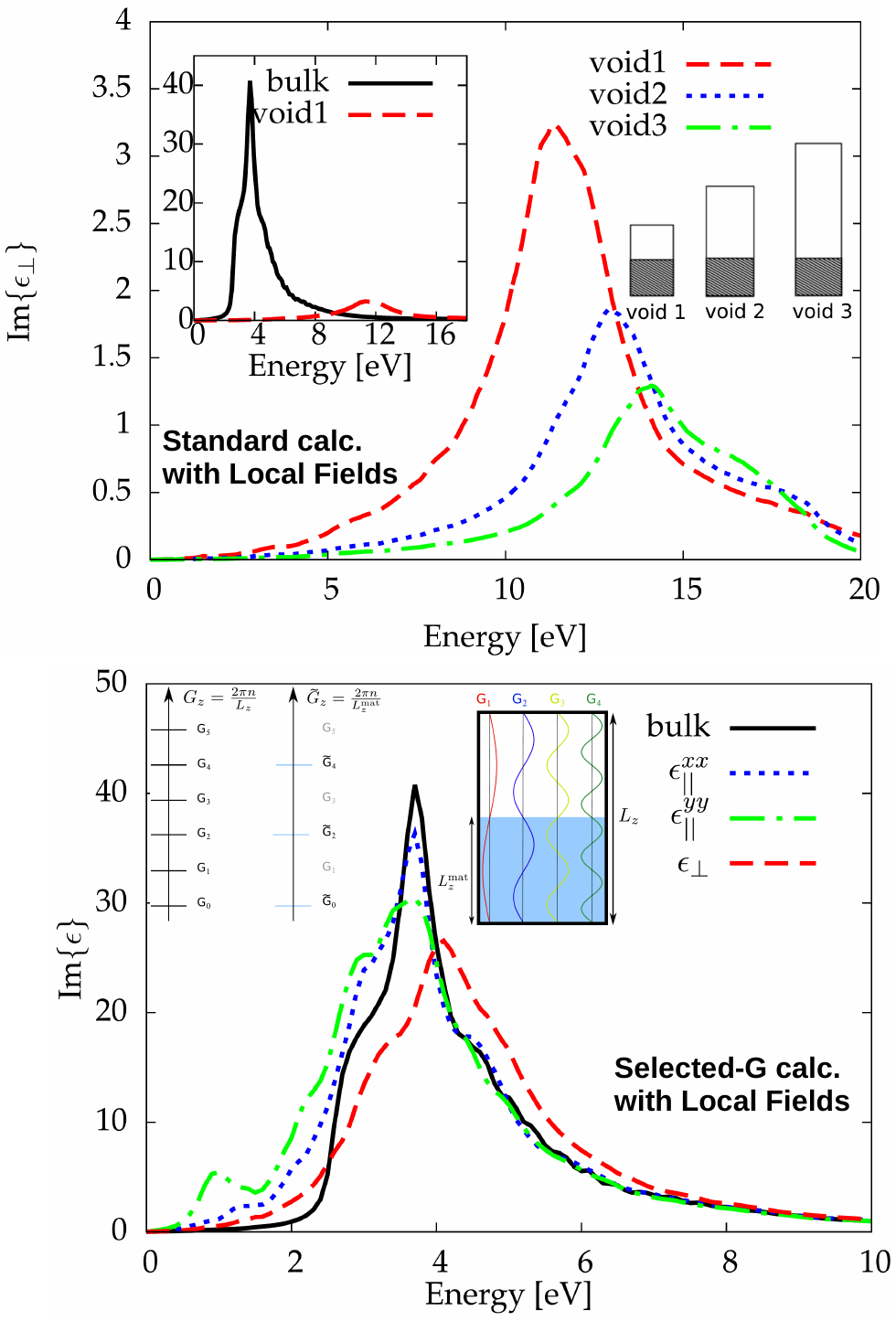
Using the TDDFT framework, we evidenced that the absorption spectrum calculated for an excitation perpendicular to the surface
correct when accounting for Local Field effects. We showed that the absorption peak was pusched up to 12 eV instead of 4 eV for the bulk counterpart, and that the energy position and the amplitude were dependent of the vacuum introduced in the supercell. We derived a modified expression for the Dyson equation of TDDFT with quantities having the periodicity of the matter instead of the supercell, and involving a new expression of the Coulomb potential. This new sheme was called Selected-G. In the limit of infinite thickness of matter, it allowed us to recover the correct absorption spectrum for the excitation perpendicular to the surface. This formalism has been successfully applied to the second harmonic generation of silicon surfaces.
Optical properties of quasi-two-dimensional objects from TDDFT
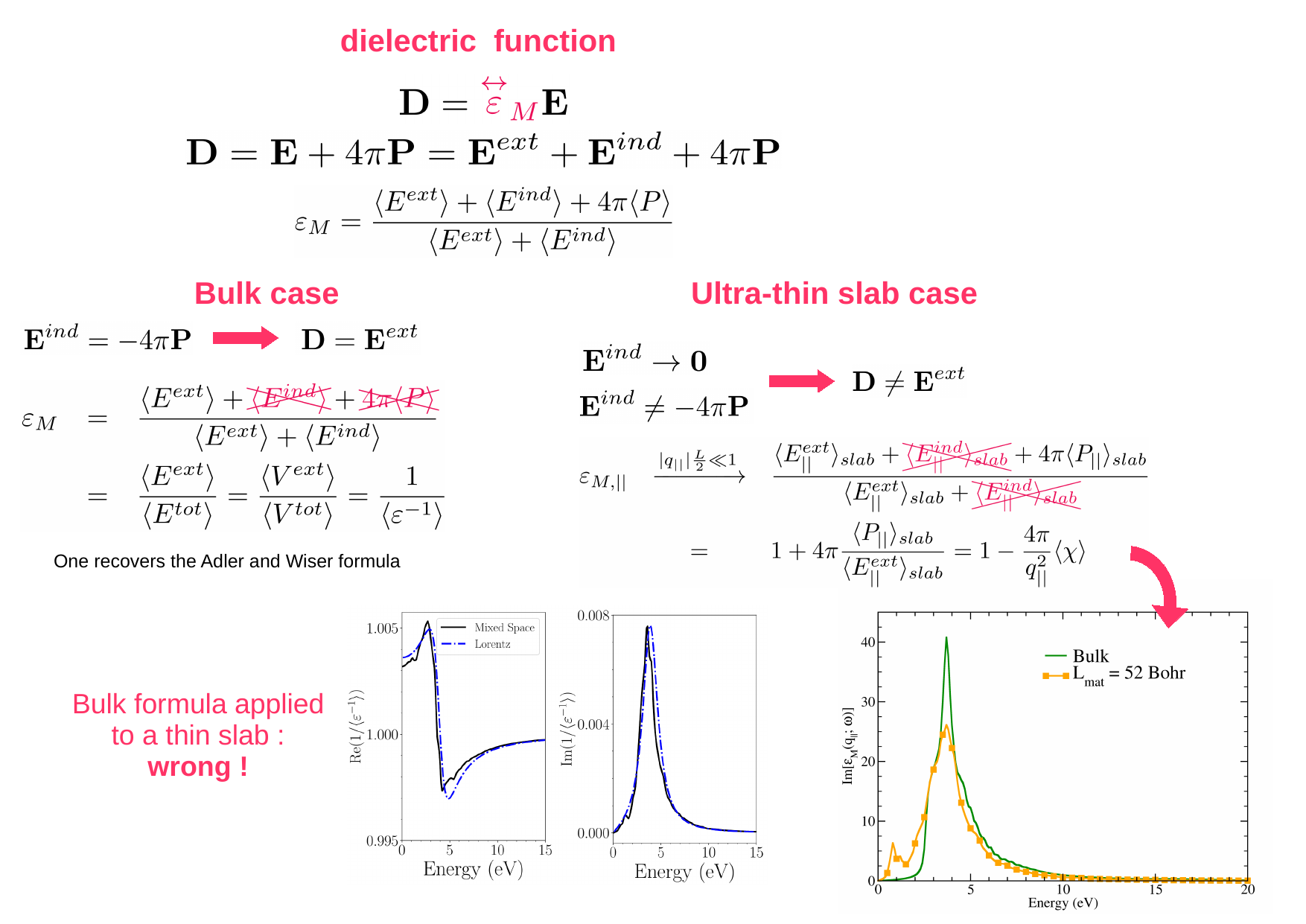
During the PhD thesis of Stefano Mazzei, associating classical electromagnetic models and TDDFT in mixed and reciprocal spaces, we evidenced that the 3D formalism is no more valid for ultra-thin materials.
A new expression for the macroscopic dielectric function
Getting insights from the classical Lorentz model, and using TDDFT in mixed space, we have demonstrated why, for an in-plane perturbation, the macroscopic average of the inverse dielectric matrix, resulting from the ratio of the total and external potentials, was a quantity almost equal to one, and could not represent to the absorption spectrum. It is a consequence of the strong reduction of the induced electric field. Coming back to the definition of the macroscopic dielectric tensor, and taking benefit from this property, we have established the formula to get the correct spectrum.
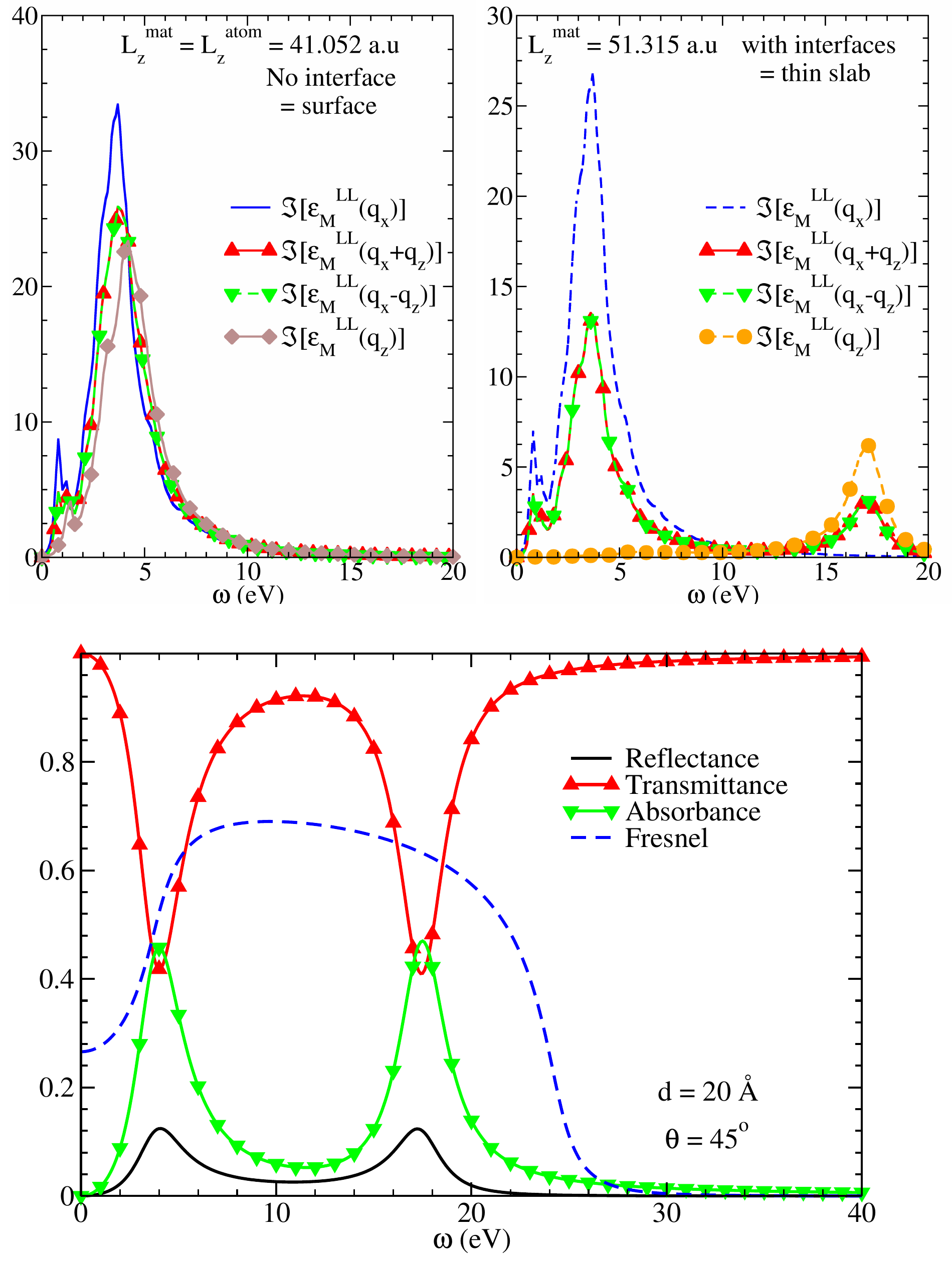
Longitudinal versus transverse dielectric functions
Calculating the response function to the total macroscopic potential, for a perturbation making an arbitrary angle with the plane of the quasi-2D object, we established that including the interfaces in the definition of the thickness of the object completely changes the nature of the properties. We showed that the response function of ultra-thin slab is a linear combination of two contributions: a resonance at the absorption frequency, associated with the in-plane projection, and a peak at the plasma frequency of the bulk counterpart for the out-of-plane one. This unexpected result was clarified with the help of the classical Airy model in the limit of the ultra-thin slab. Indeed, we evidenced that the transmittance, reflectance and absorbance exhibit the same behavior. This allows us to conclude that
i) for the out-of-plane perturbation, the LL contraction of the dielectric tensor, which reveals to be a plasmon in no more equal to the TT one, which still expected to give the absorption spectrum.
ii) the longitudinal formalism of TDDFT is still able to calculate the optical response, that is not the TT contraction of the dielectric tensor.